
炎癥(inflammation)是生物組織受到外傷、感染等損傷因子等刺激所發(fā)生的一系列以防御反應(yīng)為主的生理過程。在急性炎癥應(yīng)答過程中,短時間內(nèi)釋放的大量炎癥因子可能引發(fā)機體的劇烈損傷,進而導(dǎo)致一系列病理進程,如急性腸炎,細菌性敗血癥等。因此,炎癥過程需要被精確調(diào)控。
近日,我校生命科學(xué)學(xué)院崔雋教授團隊鑒定了一個新的組蛋白H2B的特異性去泛素化酶USP38,并揭示其通過抑制組蛋白H2B K120位點的單泛素化,及通過募集并穩(wěn)定去甲基化酶KDM5B,協(xié)同促進IL-6、IL-23a等促炎癥細胞因子啟動子附近的組蛋白H3K4的去甲基化,從表觀遺傳學(xué)水平抑制炎癥應(yīng)答的新機制。此項研究成果作為封面文章發(fā)表在Advanced Science雜志(IF=15.8)上。

圖1:Advanced Science(Volume 7, Issue 22)封面
該研究通過構(gòu)建急性敗血癥模型和結(jié)腸炎的小鼠模型,發(fā)現(xiàn)當去泛素酶USP38基因缺失時,小鼠對于細菌脂多糖 (LPS) 誘導(dǎo)的急性肺損傷以及葡聚糖酸鈉 (DSS) 誘導(dǎo)的結(jié)腸損傷具有較強的保護作用,揭示USP38對于炎癥具有重要的負向調(diào)節(jié)功能。通過轉(zhuǎn)錄組RNA-seq以及ChIP-seq等組學(xué)分析手段,該項研究發(fā)現(xiàn)USP38在炎癥應(yīng)答過程中能夠特異性的調(diào)控特異的炎性因子IL-6、IL-23等,但對于TNF影響不大。進一步的機制研究顯示USP38在炎癥刺激條件下可以定位到核小體上,通過特異性的去除組蛋白H2B的K120位點的單泛素化,連鎖抑制組蛋白H3K4的三甲基化水平,從而負向調(diào)控其下游的炎癥因子的表達。之前有研究表明組蛋白H2B的單泛素化修飾是H3K4三甲基化以及H3K79三甲基化的必要信號,促進H2B泛素化能夠促進COMPASS復(fù)合體激活,從而增強H3K4的三甲基化,但H2B泛素切割導(dǎo)致的H3K4去甲基化機制還未有報道。本項研究揭示USP38不僅能夠通過抑制H2B的泛素化負調(diào)控H3K4的三甲基化,還能夠直接結(jié)合并穩(wěn)定組蛋白H3K4的去甲基化酶KDM5B,從而進一步增強其對H3K4的去甲基化功能。
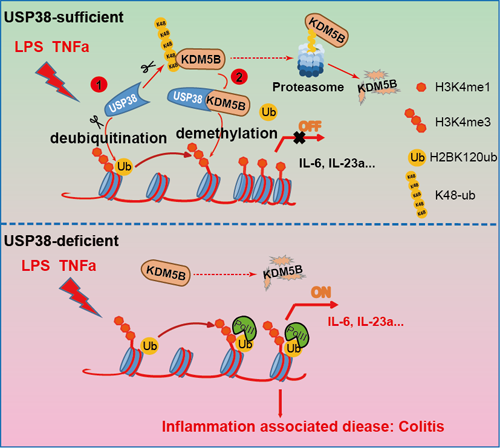
圖2:USP38-KDM5B復(fù)合物調(diào)控組蛋白泛素化和甲基化抑制炎癥應(yīng)答
綜上所述,該工作揭示了USP38-KDM5B蛋白質(zhì)復(fù)合物在炎癥應(yīng)答過程中通過連鎖抑制組蛋白泛素化與甲基化的水平,選擇性抑制炎癥因子的表達,從而負向調(diào)控炎癥發(fā)展進程的新分子機制。
我校生命科學(xué)學(xué)院崔雋教授為論文通訊作者,趙芝瑤博士為第一作者。黃軍就教授和梁普平教授團隊為該項工作的動物模型實驗研究提供了重要支持。該研究得到了國家自然科學(xué)基金等項目資助。



