
固有免疫是宿主防御入侵機體的病原微生物的第一道防線。感染病毒后,機體通過模式識別受體識別病毒的DNA或RNA,從而快速啟動固有免疫和抗病毒反應,抑制病毒的復制,進而有效清除入侵的病毒。在感知危險信號后,調控細胞抗病毒免疫的關鍵激酶TBK1(TANK-binding kinase 1)被激活,并進一步磷酸化轉錄因子IRF3,使其入核,誘導下游干擾素的產生。TBK1是固有免疫細胞抗病毒信號通路的中心節點,如果其活性不受控制,可能導致自身免疫疾病和慢性炎癥等疾病。多種翻譯后修飾參與TBK1活性的調控,包括磷酸化,泛素化,SUMO化及乙酰化。崔雋教授團隊曾揭示NLRP4-USP38軸能夠調控TBK1通過泛素-蛋白酶體途徑降解(Nature Immunology 2012; Molecular Cell 2016),然而,是否存在其他的降解系統精確調控TBK1的穩定性目前尚不明確。
細胞自噬是真核生物體內的高度保守的降解系統,能夠降解細胞內有害的蛋白聚集物、老化的細胞器和侵染機體的病原微生物等,在維持機體內穩態過程中發揮不可或缺的作用。細胞自噬能夠廣泛地參與固有免疫過程:一方面,細胞自噬可以直接降解清除入侵的病原微生物;另一方面,選擇性自噬可以通過降解抗病毒免疫網絡中的核心分子,維持免疫反應的穩態。有趣的是TBK1在自噬過程中也發揮了重要的作用,它不僅可以通過促進ULK1復合體的形成促進自噬;還可以通過調節自噬識別受體的活性,促進選擇性自噬。這些研究提示TBK1可能在細胞自噬和抗病毒免疫的crosstalk中發揮重要作用。
崔雋教授團隊發現,NEDD4能夠顯著抑制抗病毒免疫反應及促進病毒復制。NEDD4在病毒感染中結合TBK1,并顯著促進TBK1的降解,但是在無病毒侵染時,NEDD4不影響TBK1的穩定性。進一步研究發現這是由于NEDD4主要靶向磷酸化的TBK1(p-TBK1),并促使p-TBK1發生自噬降解。當TBK1的磷酸化位點(S172)突變后,NEDD4不再能夠與之結合并促進其發生自噬降解。NEDD4促使激活的TBK1在K344位點發生K27類型的泛素化修飾。選擇性自噬識別受體NDP52通過K27類型的泛素鏈識別TBK1并將其輸送至自噬溶酶體中進行降解。以往的研究表明在病毒侵染的早期,TBK1會發生K48類型的泛素修飾,進而通過泛素-蛋白酶體途徑降解。該研究進一步揭示在病毒侵染的后期,激活的TBK1主要發生K27類型的泛素修飾,使得TBK1通過選擇性自噬降解途徑進一步降解,防止TBK1被過度激活。因此,NEDD4介導的TBK1的自噬降解對于維持抗病毒免疫平衡具有重要的作用。
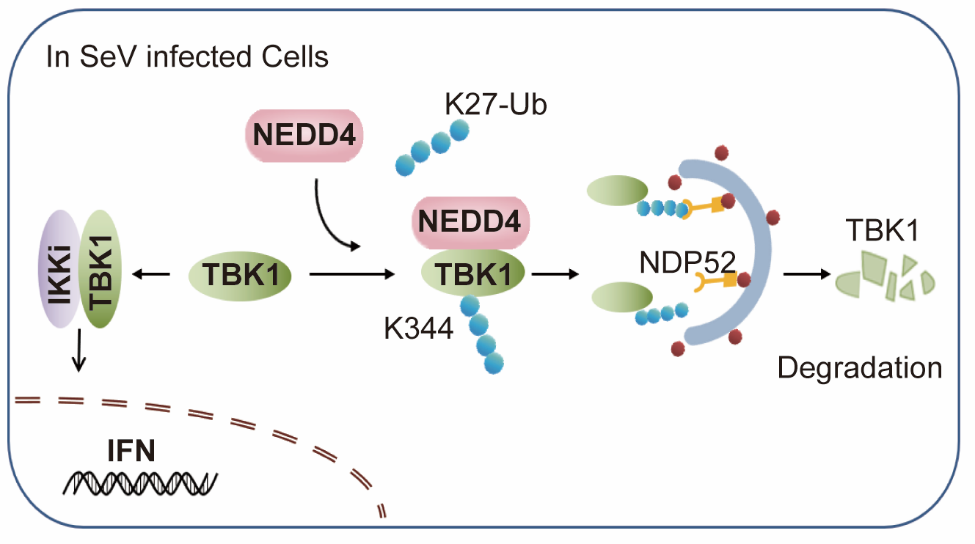
NEDD4介導的選擇性自噬通過降解TBK1維持免疫平衡的工作模式圖
該研究成果“Selective autophagy controls the stability of TBK1 via NEDD4 to balance host defense”發表在Cell Death & Differentiation雜志上。該研究揭示了NEDD4通過介導磷酸化的TBK1發生K27類型的泛素化修飾,進而使其被貨物受體NDP52識別并運輸至自噬溶酶體中降解,從而防止TBK1過度激活,維持機體免疫平衡的新機制。
我校生命科學學院崔雋教授為該論文的通訊作者,謝偉紅博士為論文第一作者。該研究得到國家自然科學基金等項目資助。



